Neurons that form part of a memory circuit are among the first brain cells to show signs of neurodegeneration in Alzheimer’s disease.
Neurodegeneration, or the gradual loss of neuron function, is one of the key features of Alzheimer’s disease. However, it doesn’t affect all parts of the brain equally.
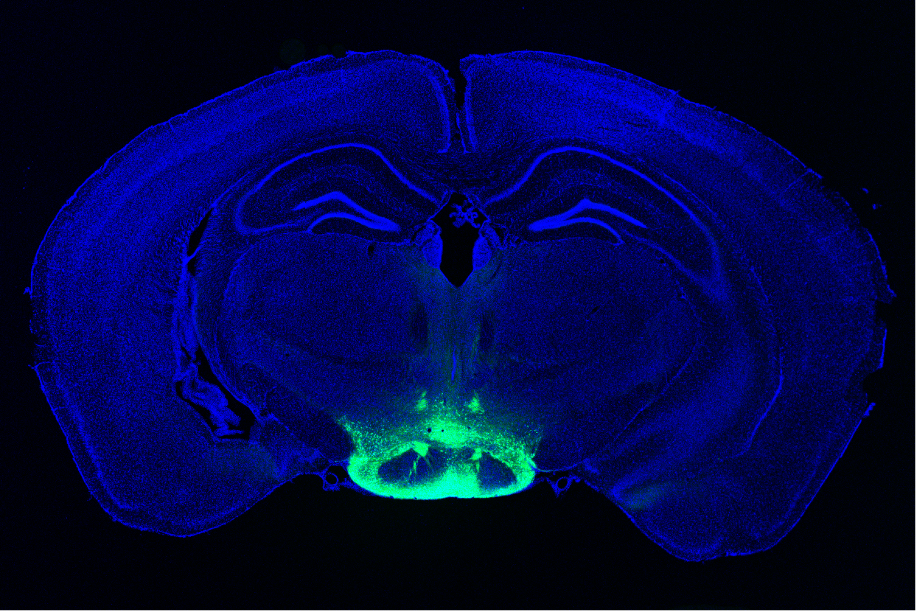
One of the first brain regions to show neurodegeneration in Alzheimer’s disease is a part of the hypothalamus called the mammillary body. In a new study, MIT researchers have identified a subset of neurons within this body that are most susceptible to neurodegeneration and hyperactivity. They also found that this damage leads to memory impairments.
The findings suggest that this region may contribute to some of the earliest symptoms of Alzheimer’s disease, making it a good target for potential new drugs to treat the disease, the researchers say.
“It is fascinating that only the lateral mammillary body neurons, not those in the medial mammillary body, become hyperactive and undergo neurodegeneration in Alzheimer’s disease,” says Li-Huei Tsai, director of MIT’s Picower Institute for Learning and Memory and the senior author of the study. Tsai also directs MIT’s Aging Brain Initiative.
In a study of mice, the researchers showed that they could reverse memory impairments caused by hyperactivity and neurodegeneration in mammillary body neurons by treating them with a drug that is now used to treat epilepsy.
Former MIT postdoc Wen-Chin (Brian) Huang and MIT graduate students Zhuyu (Verna) Peng and Mitchell Murdock are the lead authors of the paper, which appears today in Science Translational Medicine.
Predisposed to degeneration
As Alzheimer’s disease progresses, neurodegeneration occurs along with the buildup of amyloid beta plaques and misfolded Tau proteins, which form tangles in the brain. One question that remains unresolved is whether this neurodegeneration strikes indiscriminately, or if certain types of neurons are more susceptible.
“If we could identify specific molecular properties of classes of neurons that are predisposed to dysfunction and degeneration, then we would have a better understanding of neurodegeneration,” Murdock says. “This is clinically important because we could find ways to therapeutically target these vulnerable populations and potentially delay the onset of cognitive decline.”
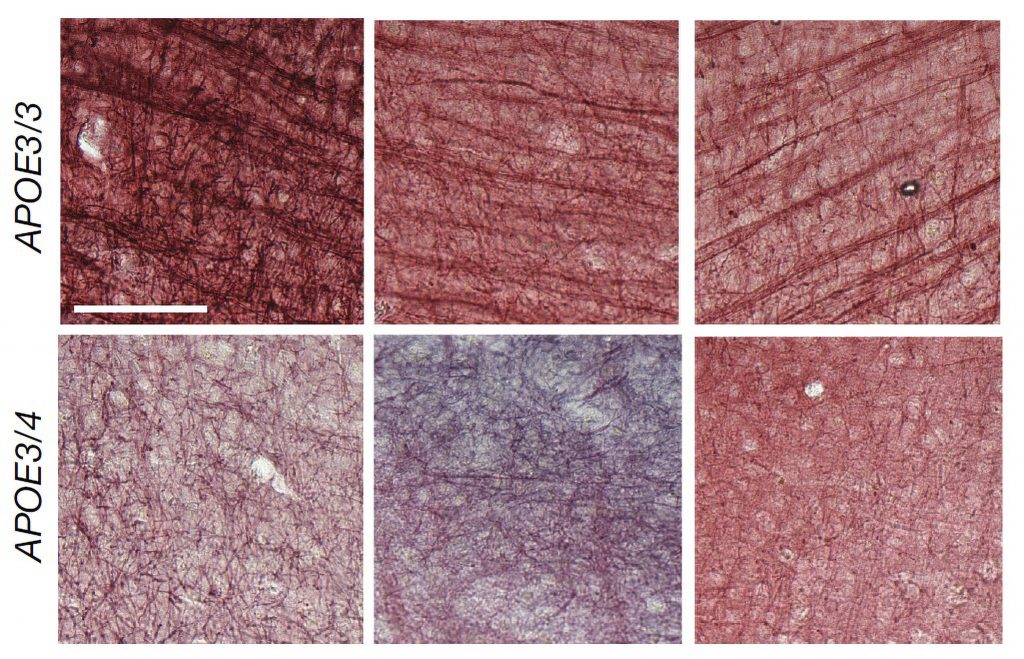
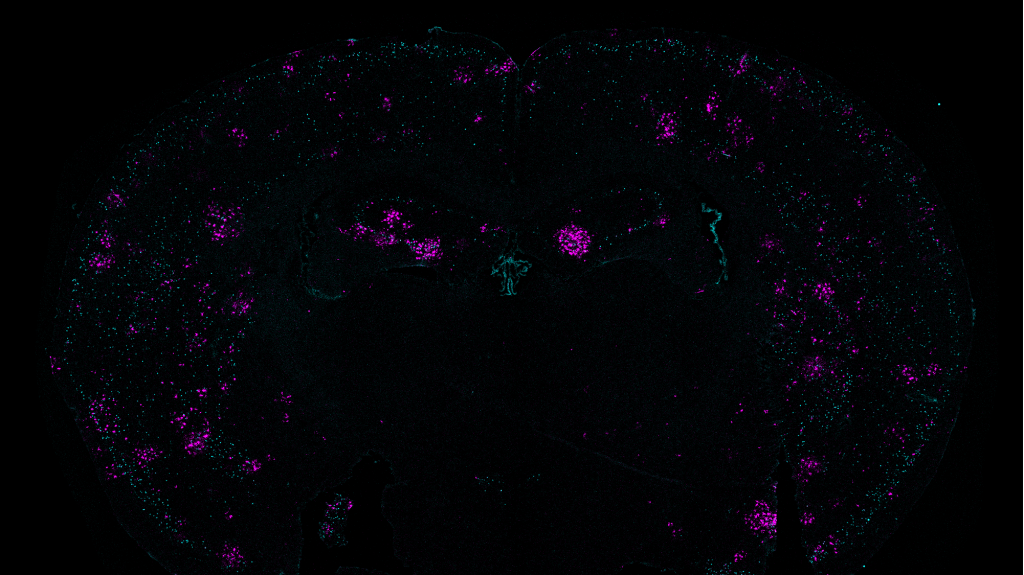
In a 2019 study using a mouse model of Alzheimer’s disease, Tsai, Huang, and others found that the mammillary bodies — a pair of structures found on the left and right underside of the hypothalamus — had the highest density of amyloid beta. These bodies are known to be involved in memory, but their exact role in normal memory and in Alzheimer’s disease is unknown.
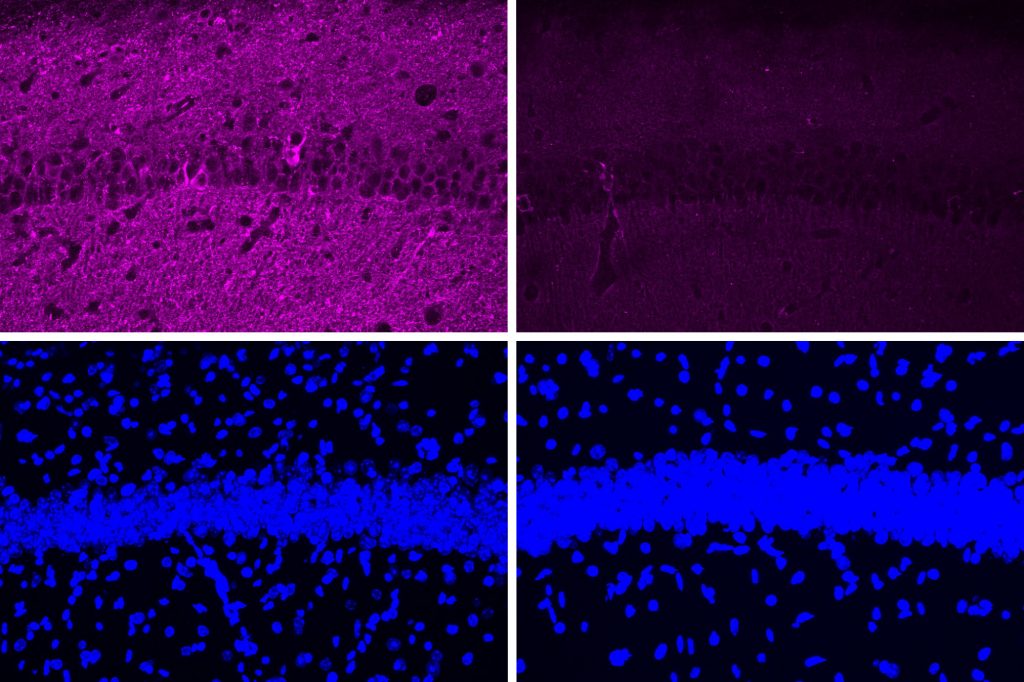
To learn more about the mammillary body’s function, the researchers used single-cell RNA-sequencing, which can reveal the genes that are active within different types of cells in a tissue sample. Using this approach, the researchers identified two major populations of neurons: one in the medial mammillary body and the other in the lateral mammillary body. In the lateral neurons, genes related to synaptic activity were very highly expressed, and the researchers also found that these neurons had higher spiking rates than medial mammillary body neurons.
Based on those differences, the researchers wondered if the lateral neurons might be more susceptible to Alzheimer’s disease. To explore that question, they studied a mouse model with five genetic mutations linked to early-onset Alzheimer’s in humans. The researchers found that these mice showed much more hyperactivity in lateral mammillary body neurons than healthy mice. However, the medial mammillary body neurons in healthy mice and the Alzheimer’s model did not show any such differences.
The researchers found that this hyperactivity emerged very early — around two months of age (the equivalent of a young human adult), before amyloid plaques begin to develop. The lateral neurons became even more hyperactive as the mice aged, and these neurons were also more susceptible to neurodegeneration than the medial neurons.
“We think the hyperactivity is related to dysfunction in memory circuits and is also related to a cellular progression that might lead to neuronal death,” Murdock says.
The Alzheimer’s mouse model showed impairments in forming new memories, but when the researchers treated the mice with a drug that reduces neuronal hyperactivity, their performance on memory tasks was significantly improved. This drug, known as levetiracetam, is used to treat epileptic seizures and is also in clinical trials to treat epileptiform activity — hyperexcitability in the cortex, which increases the risk of seizures — in Alzheimer’s patients.
Comparing mice and humans
The researchers also studied human brain tissue from the Religious Orders Study/Memory and Aging Project (ROSMAP), a longitudinal study that has tracked memory, motor, and other age-related issues in older people since 1994. Using single-cell RNA-sequencing of mammillary body tissue from people with and without Alzheimer’s disease, the researchers found two clusters of neurons that correspond to the lateral and medial mammillary body neurons they found in mice.
Similar to the mouse studies, the researchers also found signatures of hyperactivity in the lateral mammillary bodies from Alzheimer’s tissue samples, including overexpression of genes that encode potassium and sodium channels. In those samples, they also found higher levels of neurodegeneration in the lateral neuron cluster, compared to the medial cluster.
Other studies of Alzheimer’s patients have found a loss of volume of the mammillary body early in the disease, along with deposition of plaques and altered synaptic structure. All of these findings suggest that the mammillary body could make a good target for potential drugs that could slow down the progression of Alzheimer’s disease, the researchers say.
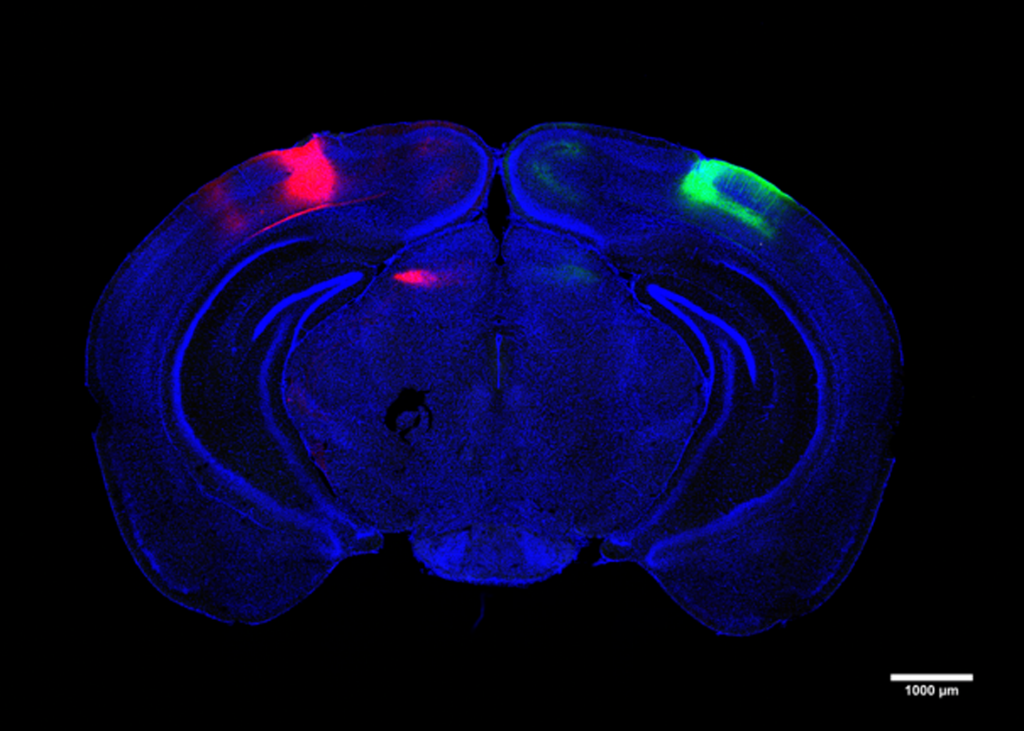
Tsai’s lab is now working on further defining how the lateral neurons of the mammillary body are connected to other parts of the brain, to figure out how it forms memory circuits. The researchers also hope to learn more about what properties of the lateral neurons of the mammillary body make them more vulnerable to neurodegeneration and amyloid deposition.
The research was funded by The JPB Foundation, the Carol and Gene Ludwig Family Foundation, and the U.S. National Institutes of Health.
–From MIT News