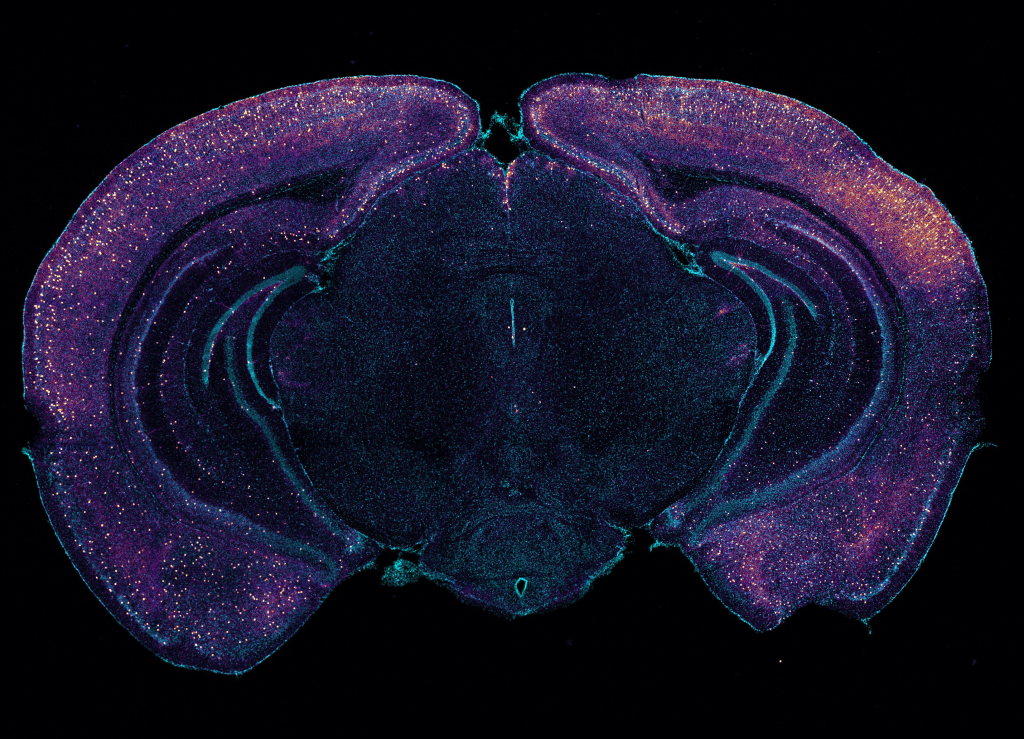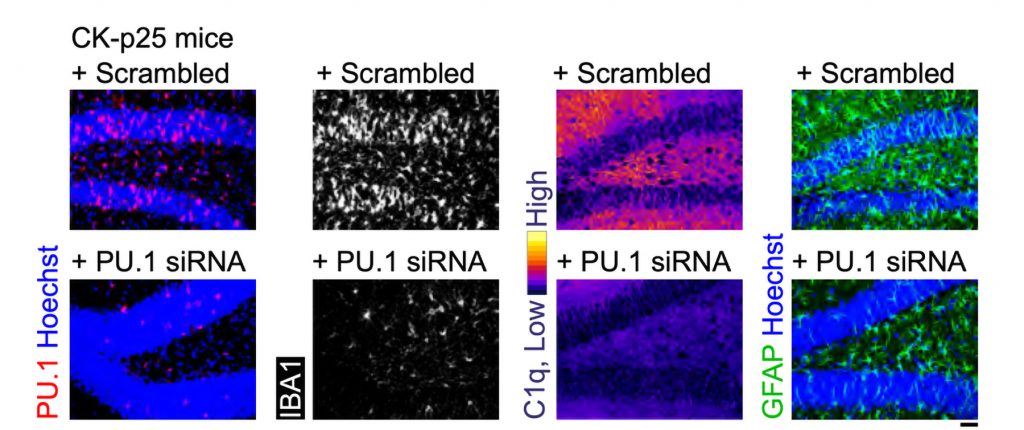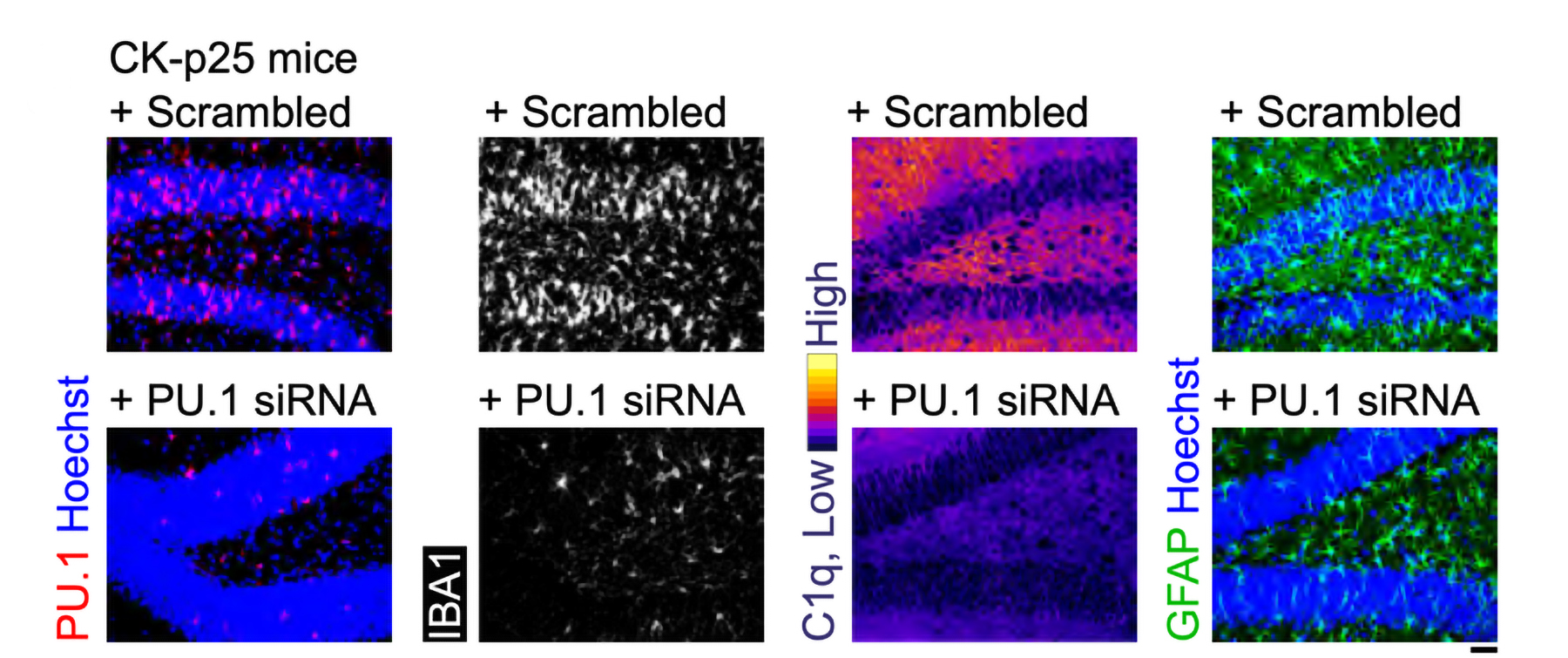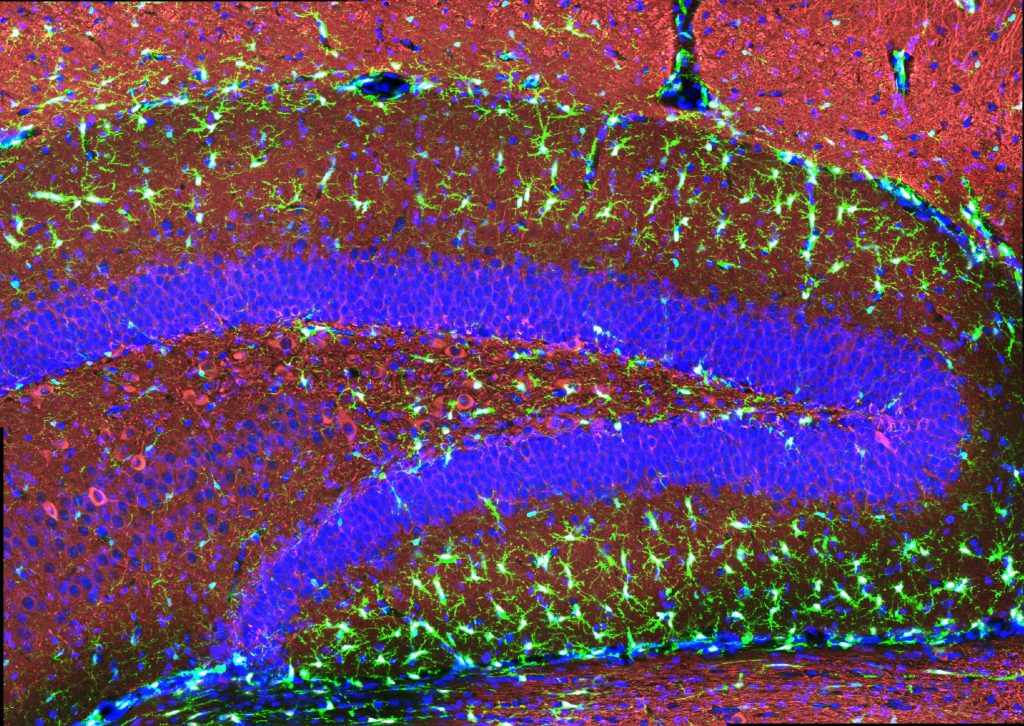
MIT scientists report that gamma frequency light and sound stimulation preserves myelination in mouse models and reveal molecular mechanisms that may underlie the benefit.
Early-stage trials in Alzheimer’s disease patients and studies in mouse models of the disease have suggested positive impacts on pathology and symptoms from exposure to light and sound presented at the “gamma” band frequency of 40 Hz. A new study zeroes in on how 40Hz sensory stimulation helps to sustain an essential process in which the signal-sending branches of neurons, called axons, are wrapped in a fatty insulation called myelin. Often called the brain’s “white matter,” myelin protects axons and insures better electrical signal transmission in brain circuits.
“Previous publications from our lab have mainly focused on neuronal protection,” said Li-Huei Tsai, Picower Professor in The Picower Institute for Learning and Memory and the Department of Brain and Cognitive Sciences at MIT and senior author of the new study in Nature Communications. Tsai also lead’s MIT’s Aging Brain Initiative. “But this study shows that it’s not just the gray matter, but also the white matter that’s protected by this method.”
This year Cognito Therapeutics, the spin-off company that licensed MIT’s sensory stimulation technology, published phase II human trial results in the Journal of Alzheimer’s Disease indicating that 40Hz light and sound stimulation significantly slowed the loss of myelin in volunteers with Alzheimer’s. Also this year Tsai’s lab published a study showing that gamma sensory stimulation helped mice withstand neurological effects of chemotherapy medicines, including by preserving myelin. In the new study, members of Tsai’s lab led by former postdoc Daniela Rodrigues Amorim used a common mouse model of myelin loss—a diet with the chemical cuprizone— to explore how sensory stimulation preserves myelination.
Amorim and Tsai’s team found that 40Hz light and sound not only preserved myelination in the brains of cuprizone-exposed mice, it also appeared to protect oligodendrocytes (the cells that myelinate neural axons), sustain the electrical performance of neurons, and preserve a key marker of axon structural integrity. When the team looked into the molecular underpinnings of these benefits, they found clear signs of specific mechanisms including preservation of neural circuit connections called synapses; a reduction in a cause of oligodendrocyte death called “ferroptosis;” reduced inflammation; and an increase in the ability of microglia brain cells to clean up myelin damage so that new myelin could be restored.
“Gamma stimulation promotes a healthy environment,” said Amorim who is now a Marie Curie Fellow at the University of Galway in Ireland. “There are several ways we are seeing different effects.”
The findings suggest that gamma sensory stimulation may help not only Alzheimer’s disease patients but also people battling other diseases involving myelin loss, such as multiple sclerosis, the authors wrote in the study.
Maintaining myelin
To conduct the study, Tsai and Amorim’s team fed some male mice a diet with cuprizone and gave other male mice a normal diet for six weeks. Halfway into that period, when cuprizone is known to begin causing its most acute effects on myelination, they exposed some mice from each group to gamma sensory stimulation for the remaining three weeks. In this way they had four groups: completely unaffected mice, mice that received no cuprizone but did get gamma stimulation, mice that received cuprizone and constant (but not 40Hz) light and sound as a control, and mice that received cuprizone and also gamma stimulation.
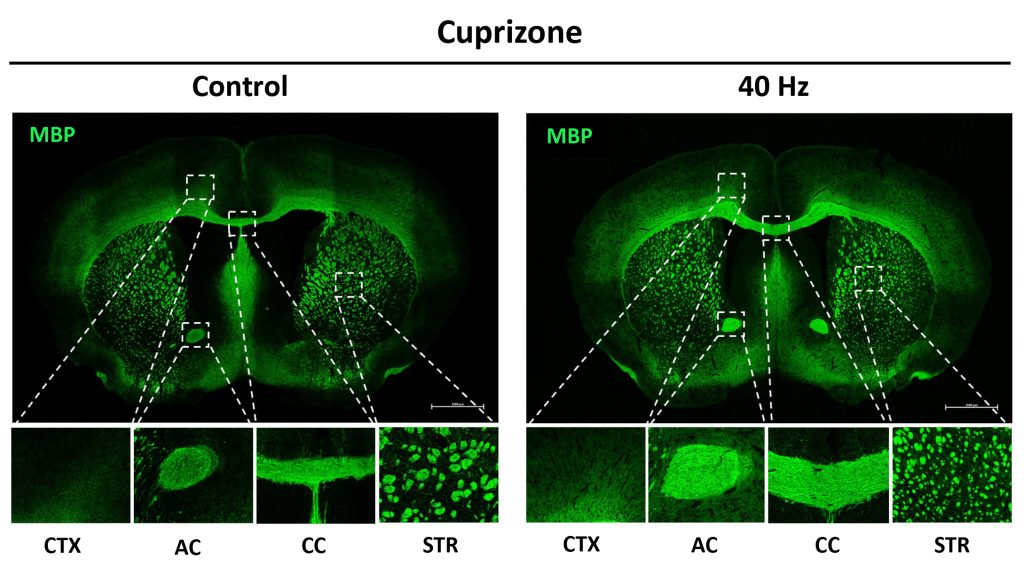
After the six weeks elapsed, the scientists measured signs of myelination throughout the brains of the mice in each group. Mice that weren’t fed cuprizone maintained healthy levels, as expected. Mice that were fed cuprizone and didn’t receive 40Hz gamma sensory stimulation showed drastic levels of myelin loss. Cuprizone-fed mice that received 40Hz stimulation retained significantly more myelin, rivaling the health of mice never fed cuprizone by some, but not all, measures.
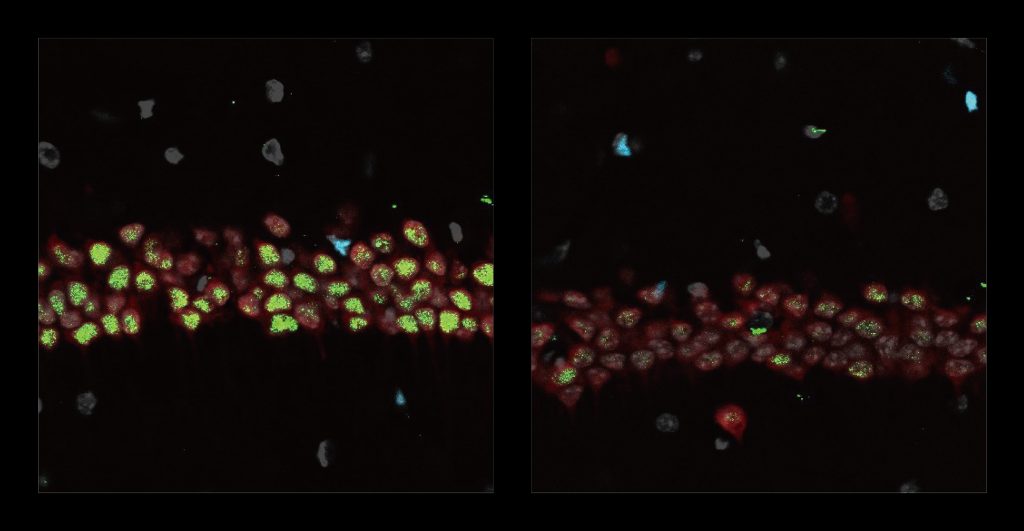
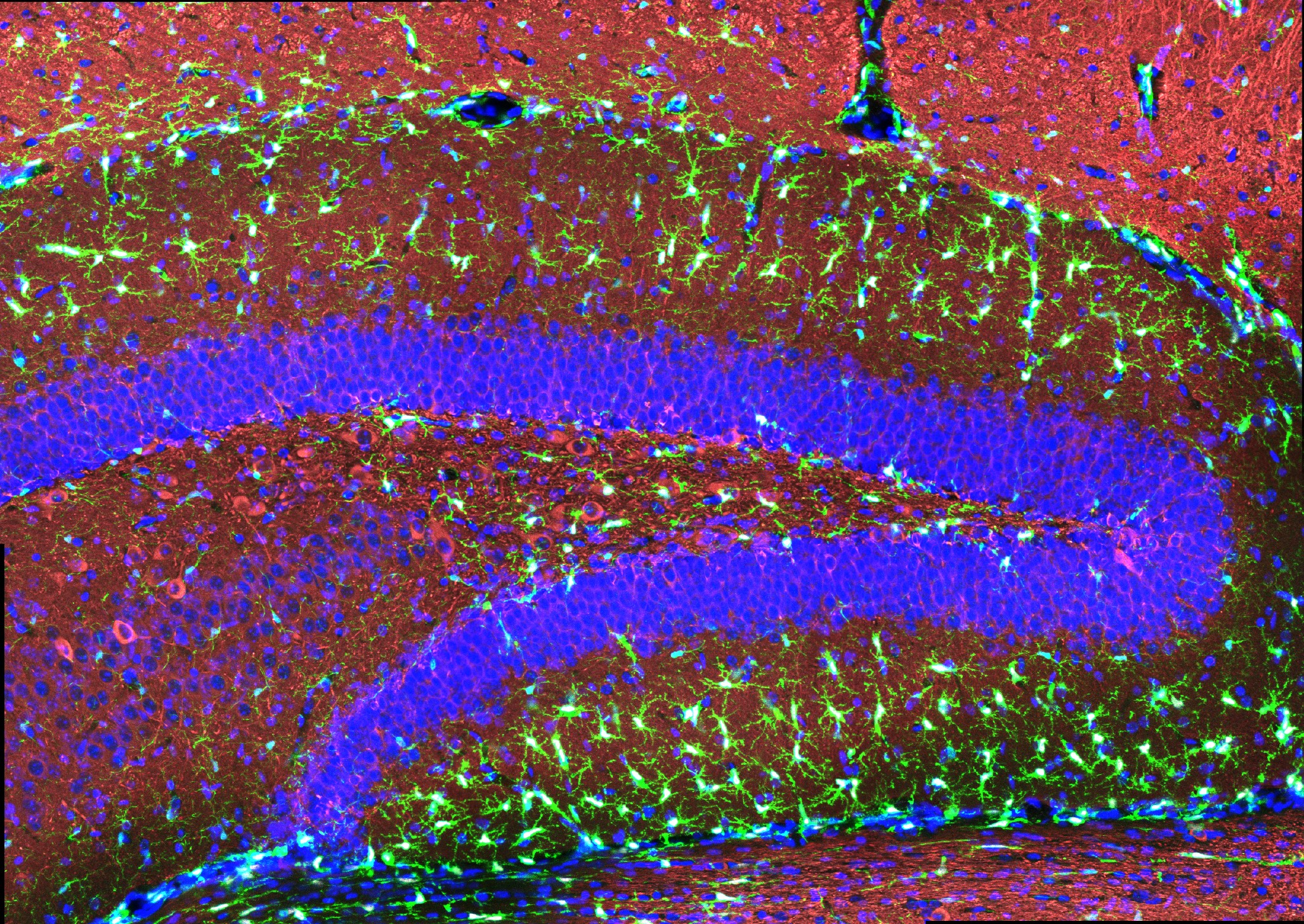
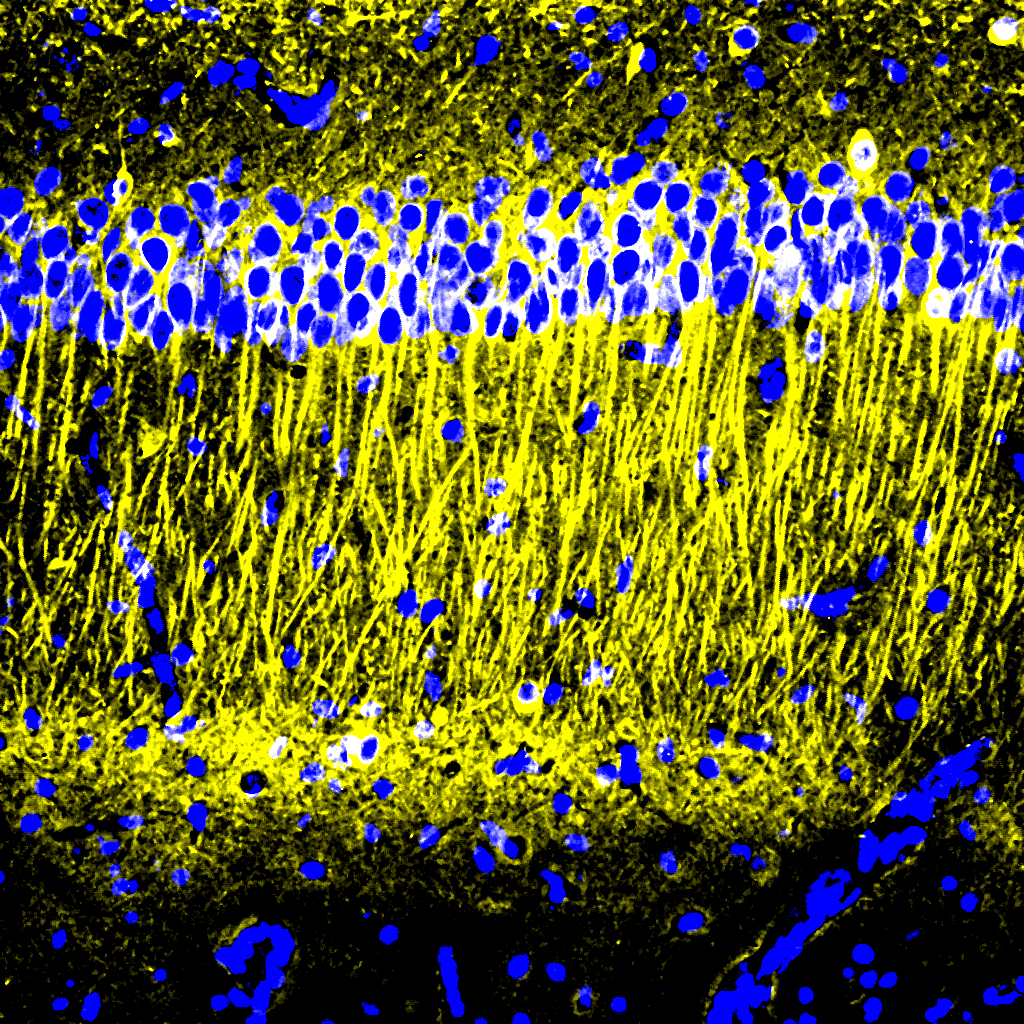
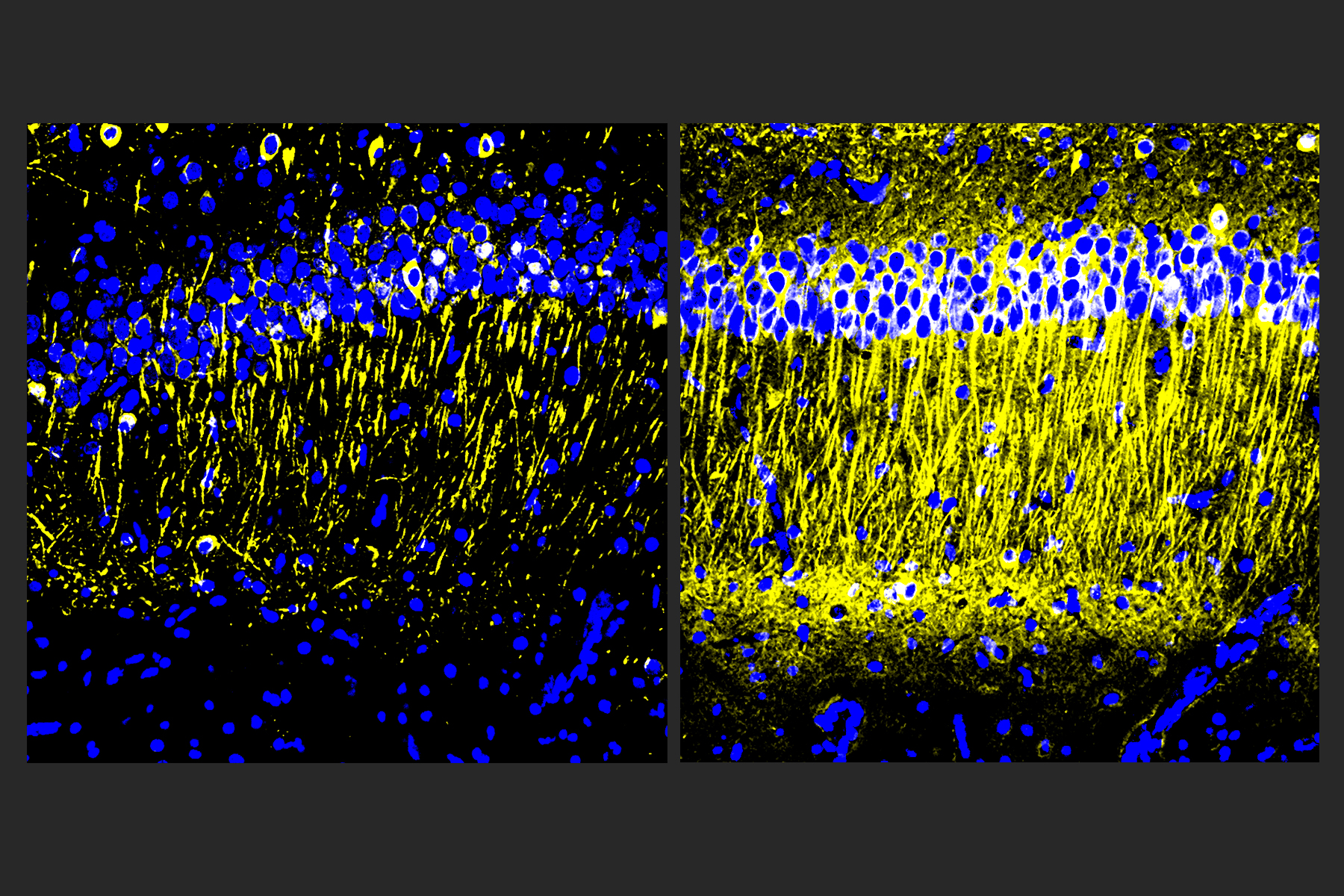
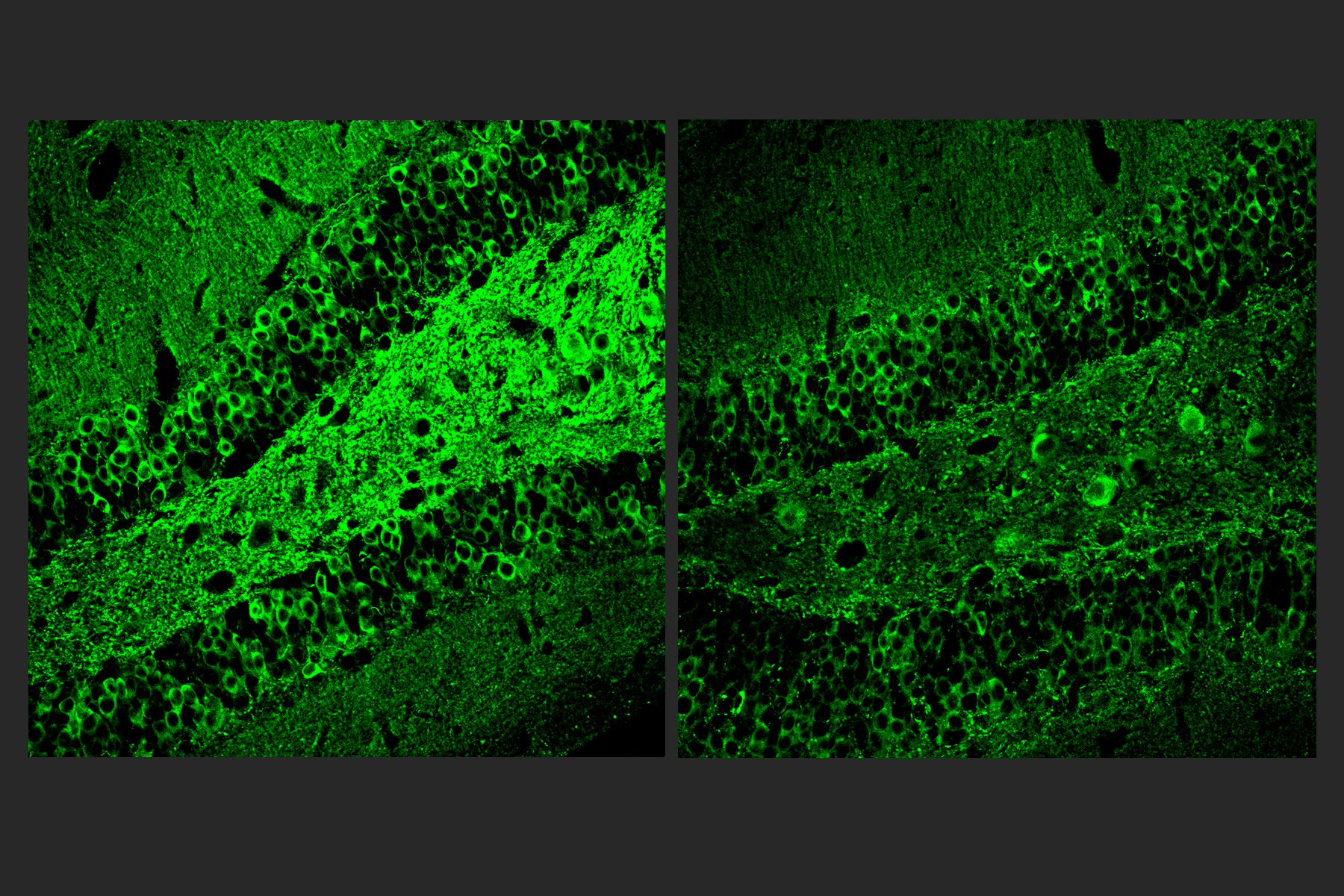
The researchers also looked at numbers of oligodendrocytes to see if they survived better with sensory stimulation. Several measures revealed that in mice fed cuprizone, oligodendrocytes in the corpus callosum region of the brain (a key point for the transit of neural signals because it connects the brain’s hemispheres) were markedly reduced. But in mice fed cuprizone and also treated with gamma stimulation, the number of cells were much closer to healthy levels.
Electrophysiological tests among neural axons in the corpus callosum showed that gamma sensory stimulation was associated with improved electrical performance in cuprizone-fed mice who received gamma stimulation compared to cuprizone-fed mice left untreated by 40Hz stimulation. And when researchers looked in the anterior cingulate cortex region of the brain, they saw that MAP2, a protein that signals the structural integrity of axons, was much better preserved in mice that received cuprizone and gamma stimulation compared to cuprizone-fed mice who did not.
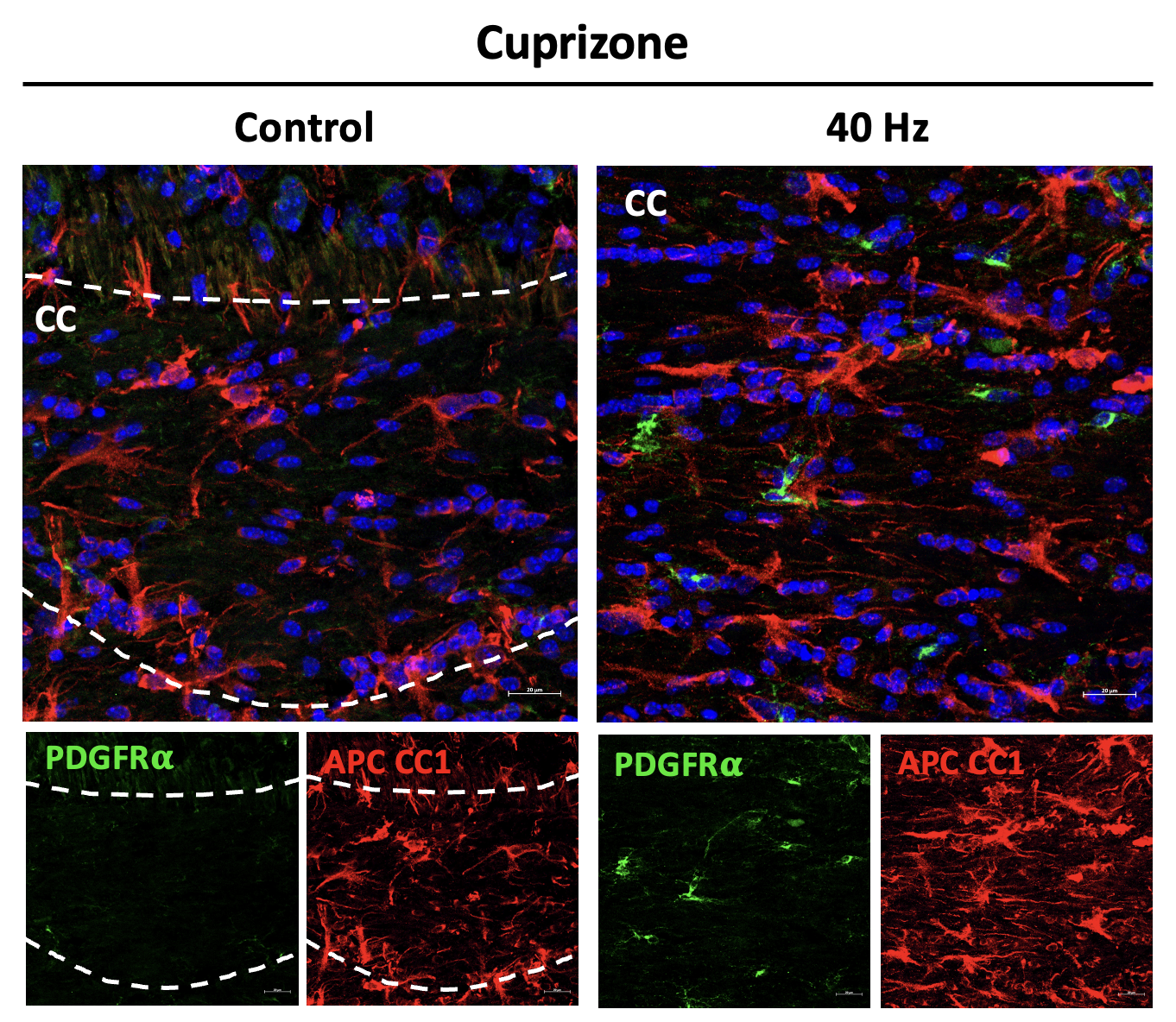
Molecular mechanisms
A key goal of the study was to identify possible ways in which 40Hz sensory stimulation may protect myelin.
To find out, the researchers conducted a sweeping assessment of protein expression in each mouse group and identified which proteins were differentially expressed based on cuprizone diet and exposure to gamma frequency stimulation. The analysis revealed distinct sets of effects between the cuprizone mice exposed to control stimulation and cuprizone-plus-gamma mice.
A highlight of one set of effects was the increase in MAP2 in gamma-treated cuprizone-fed mice. A highlight of another set was that cuprizone mice who received control stimulation showed a substantial deficit in expression of proteins associated with synapses. The gamma-treated cuprizone-fed mice did not show any significant loss, mirroring results in a 2019 Alzheimer’s 40Hz study that showed synaptic preservation. This result is important, the researchers wrote, because neural circuit activity, which depends on maintaining synapses, is associated with preserving myelin. They confirmed the protein expression results by looking directly at brain tissues.
Another set of protein expression results hinted at another important mechanism: ferroptosis. This phenomenon, in which errant metabolism of iron leads to a lethal buildup of reactive oxygen species in cells, is a known problem for oligodendrocytes in the cuprizone mouse model. Among the signs was an increase in cuprizone-fed, control stimulation mice in expression of the protein HMGB1, which is a marker of ferroptosis-associated damage that triggers an inflammatory response. Gamma stimulation, however, reduced levels of HMGB1.
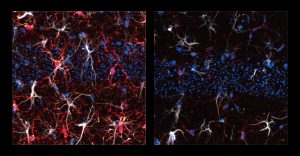
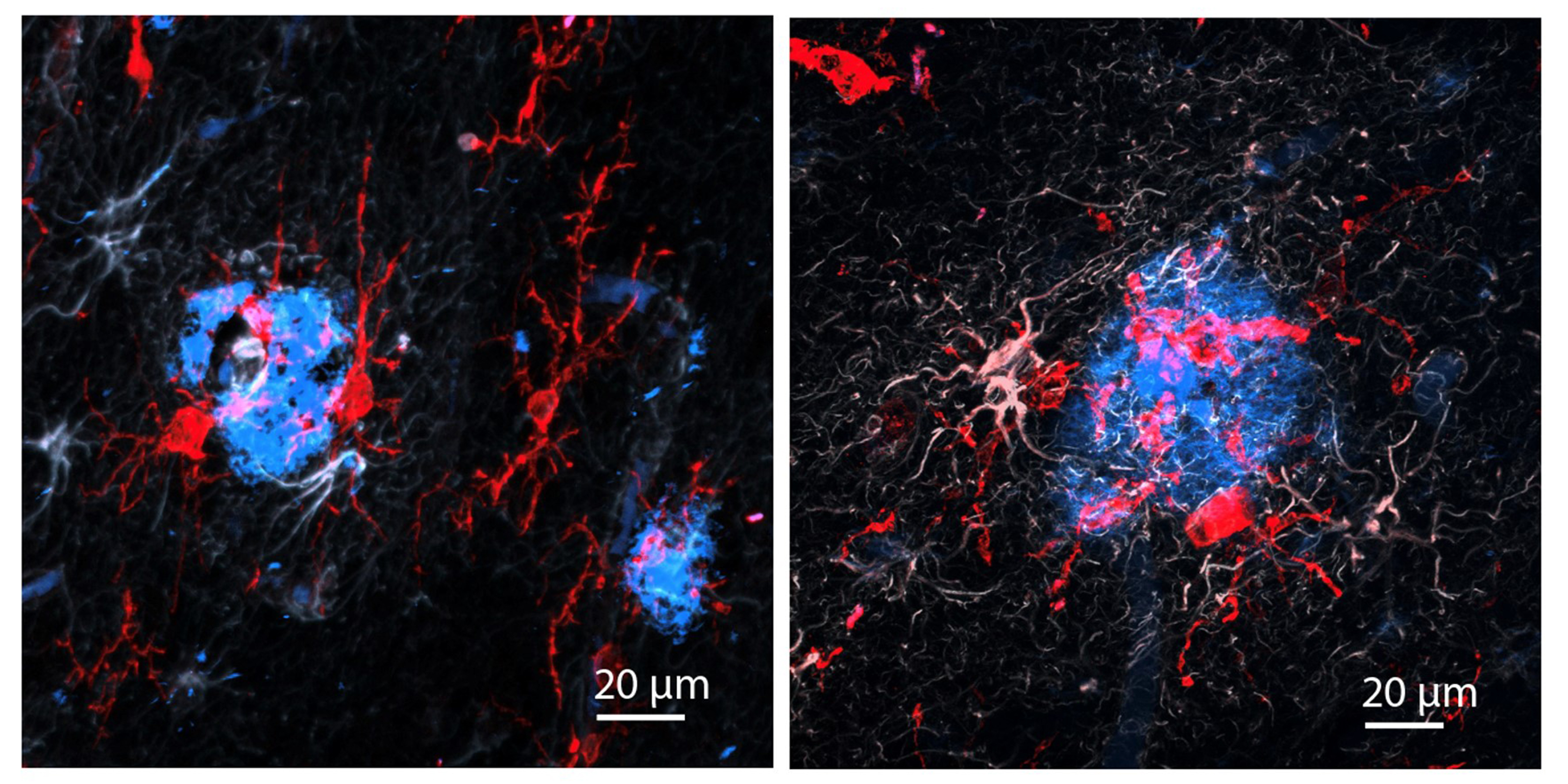
Looking more deeply at the cellular and molecular response to cuprizone demyelination and the effects of gamma stimulation, the team assessed gene expression using single-cell RNA sequencing technology. They found that astrocytes and microglia became very inflammatory in cuprizone-control mice but gamma stimulation calmed that response. Fewer cells became inflammatory and direct observations of tissue showed that microglia became more proficient at clearing away myelin debris, a key step in effecting repairs.
The team also learned more about how oligodendrocytes in cuprizone-fed mice exposed to 40Hz sensory stimulation managed to survive better. Expression of protective proteins such as HSP70 increased and as did expression of GPX4, a master regulator of processes that constrain ferroptosis.
In addition to Amorim and Tsai, the paper’s other authors are Lorenzo Bozzelli, TaeHyun Kim, Liwang Liu, Oliver Gibson, Cheng-Yi Yang, Mitch Murdock, Fabiola Galiana-Meléndez, Brooke Schatz, Alexis Davison, Md Rezaul Islam, Dong Shin Park, Ravikiran M. Raju, Fatema Abdurrob, Alissa J. Nelson, Jian Min Ren, Vicky Yang and Matthew P. Stokes.
Fundacion Bancaria la Caixa, The JPB Foundation, The Picower Institute for Learning and Memory, the Carol and Gene Ludwig Family Foundation, Lester A. Gimpelson, Eduardo Eurnekian, The Dolby Family, Kathy and Miguel Octavio, the Marc Haas Foundation, Ben Lenail and Laurie Yoler, and the National Institutes of Health provided funding for the study.